XIV.8 II. Hauptsatz der Wärmelehre und Entropie
Die Mannigfaltigkeit der möglichen thermodynamischen Prozesse wird durch den I. Hauptsatz beschränkt, weil dort nur solche Prozesse zugelassen sind, die dem Prinzip der Erhaltung der Energie unterliegen. In solchen Prozessen kann Energie weder erzeugt noch vernichtet werden, sondern nur von einer Form in die andere umgewandelt werden. Wir hatten bereits angesprochen, daß ein daraus resultierender Wirkungsgrad einer periodisch arbeitenden Maschine von 100% in der Realität jedoch nicht zu bewerkstelligen ist. Betrachtet man noch andere in der Natur und in den technischen Anlagen ablaufenden Prozesse, so sieht man, daß viele Prozesse nicht vorkommen, obwohl sie mit dem I. Hauptsatz verträglich sind. Hier einige Beispiele:
Es werden zwei Körper (Systeme) von verschiedener Temperatur in thermischen Kontakt gebracht. Dann fließt eine Wärmemenge solange vom wärmeren Körper zum kälteren, bis sich die Temperaturen beider Körper angeglichen haben. Die innere Energie des kälteren Körpers erhöht sich dabei um die Wärmemenge, die von dem wärmeren zugeflossen ist. Um diesen Betrag erniedrigt sich die innere Energie des wärmeren Körpers. Der umgekehrte Prozeß, bei dem vom kälteren Körper eine Wärmemenge zum wärmeren fließt und somit die Temperaturdifferenz erhöht, oder daß nach dem Temperaturausgleich sich von selbst wieder ein Temperaturunterschied zwischen den beiden Körpern einstellen würde, steht im Einklang mit dem I. Hauptsatz. Diese Prozesse sind aber noch nie beobachtet worden.
Ein weiteres Beispiel soll der Versuch von Gay-Lussac sein. Bei diesem Versuch, den wir bereits betrachtet haben, expandiert Gas frei in einen größeren Raum bzw. größeres Volumen. Hierbei wird keine Arbeit verrichtet und es findet auch kein Wärmeaustausch mit der Umgebung statt, so daß die innere Energie konstant bleibt. Die Umkehrung dieses Prozesses, d.h. daß sich das Gas von selbst wieder auf das Ausgangsvolumen zurückzieht und somit den Anfangszustand einnehmen würde, steht nicht im Widerspruch zum I. Hauptsatz. Ein solcher Vorgang tritt aber ebenfalls nie auf. Ähnliches gilt auch für die Diffusion von zwei unterschiedlichen Gasen.
Diese Beispiele zeigen, daß der I. Hauptsatz keineswegs ausreicht, um die in der Natur wirklich vorkommenden Prozesse eindeutig zu bestimmen und deren Richtung festzulegen. Die hierfür erforderlichen Bedingungen sind Inhalt des II. Hauptsatzes der Thermodynamik.
Es gibt kein Perpetuum mobile 2. Art.
Diese vorweggenommene Aussage ist bereits eine Formulierung des II. Hauptsatzes:
0. Formulierung des II. Hauptsatzes: Es gibt kein Perpetuum mobile 2. Art. |
Eine ausführliche Formulierung, welche die Definition eines Perpetuum mobile 2.Art mit einschließt, wurde von z.B. Lord Kelvin, R. Clausius(1822 - 1888) und M. Planck (1858 - 1947) aufgestellt. Als Beispiel geben wir Oswalds Formulierung an:
I. Formulierung des II. Hauptsatzes: Oswalds Formulierung "Es gibt keine periodisch arbeitende Maschine, die nichts anderes bewirkt als Erzeugung mechanischer Arbeit und Abkühlung eines Wärmereservoirs." |
Entropiesatz:
"Jedes thermodynamische System besitzt eine Zustandsgröße, die Entropie S. Man berechnet sie, indem man das System aus einem willkürlich gewählten Anfangszustand in den jeweiligen Zustand des Systems durch eine Folge von Gleichgewichtszuständen überführt, die hierbei schrittweise reversibel zugeführte Wärme d Qrev bestimmt, letztere durch die absolute Temperatur dividiert und sämtliche Quotienten aufsummiert. Bei wirklichen Zustandsänderungen eines abgeschlossenen Systems kann die Entropie nur zunehmen (irreversible Änderung) oder konstant bleiben (reversible Änderung)." |
Definition XIV.8: Die Entropieänderung D S eines Körpers (Systems) beim Übergang von einem Zustand in einen anderen ist gegeben durch das Integral |
Der Index rev besagt, daß der Weg zwischen den Zuständen reversibel durchlaufbar sein muß.
Die Entropie S ist ein Maß für vergeudete Arbeit oder erreichte Irreversibilität eines Zustandes.
Mit dieser neu definierten Größe können wir den II.Hauptsatz formulieren als:
II. Formulierung des II. Hauptsatzes:
Die Entropie eines Systems kann bei beliebigen Vorgängen immer nur zunehmen: |
Zusammenfassend können wir aus diesen Beispielen festhalten, daß es Prozesse gibt, die nicht umkehrbar sind. Noch einige weitere Beispiele auch aus dem Bereich der Mechanik für reversible und irreversible Prozesse sind in einer
![]() Tabelle aufgeführt.
Tabelle aufgeführt.
Diesen Beweis führen wir in zwei Schritten:
1. Schritt: Die Entropie ist eine Zustandsgröße
Bei der Definition der thermodynamischen Temperatur hatten wir errechnet, daß bei einem Carnot-Prozess die Beziehung gilt
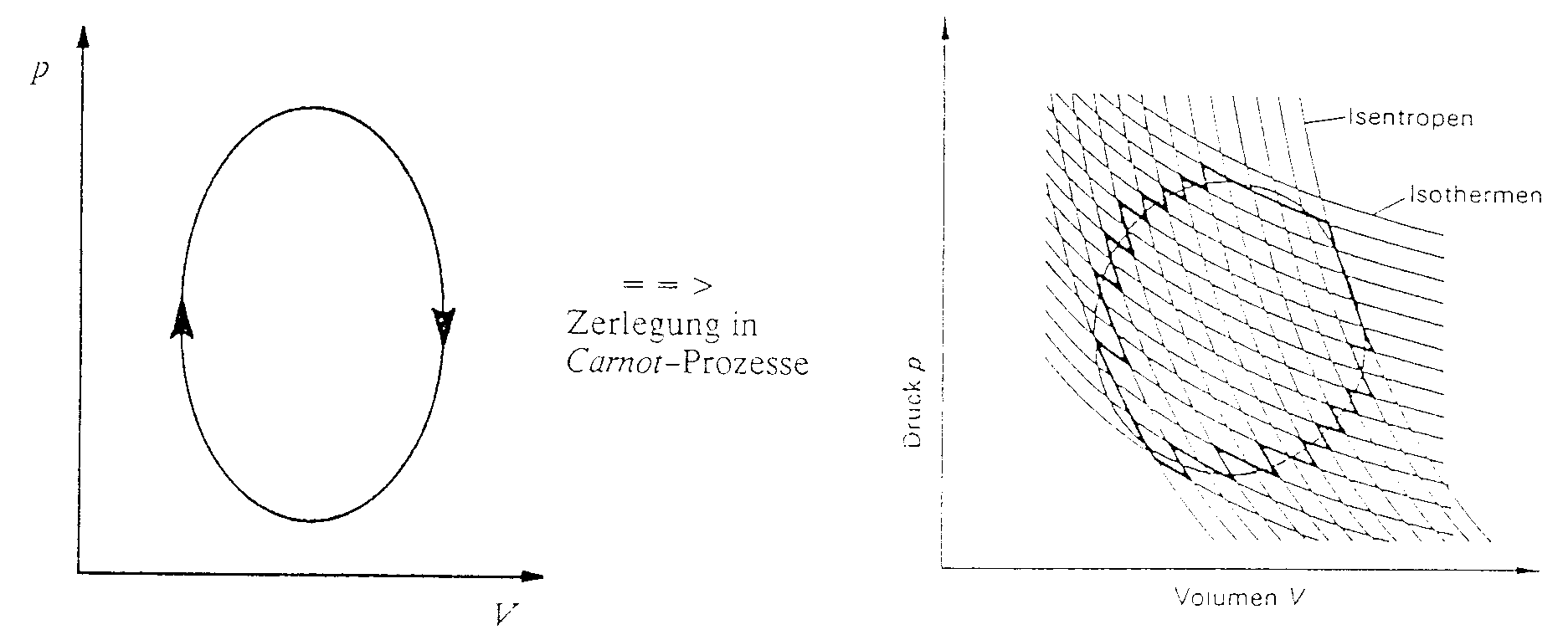
Im Prinzip kann jeder beliebige reversible Kreisprozeß durch unendlich viele differentiell schmale Carnot-Prozesse ersetzt werden.
Dann muß bei einer Zerlegung in n Teilprozesse gelten:
da bei reversiblen Prozessen die Änderung der Entropie ![]() ist.
ist.
Gehen wir zu einer beliebig feinen Zerlegung über, die den betrachteten Gesamtprozeß beliebig genau annähert, dann müssen wir die Summe durch ein Ringintegral ersetzen, d.h. wir machen den Grenzübergang:
Der Index rev soll daran erinnern, daß die Beziehung nur für reversible Prozesse gilt. Da die obige Beziehung über eine geschlossene Kurve Null ist, deutet sie auf eine neue Zustandsgröße hin. Diese Zustandsgröße bezeichnet man nach R. Claudius als Entropie S. Ihr Differential ist definiert als:
Dies ist die bereits bekannte thermodynamische Definition der Entropie. Man nennt sie auch Änderung der Entropie.
Aus dem Ergebnis für einen reversiblen Kreisprozeß
folgt nun, daß
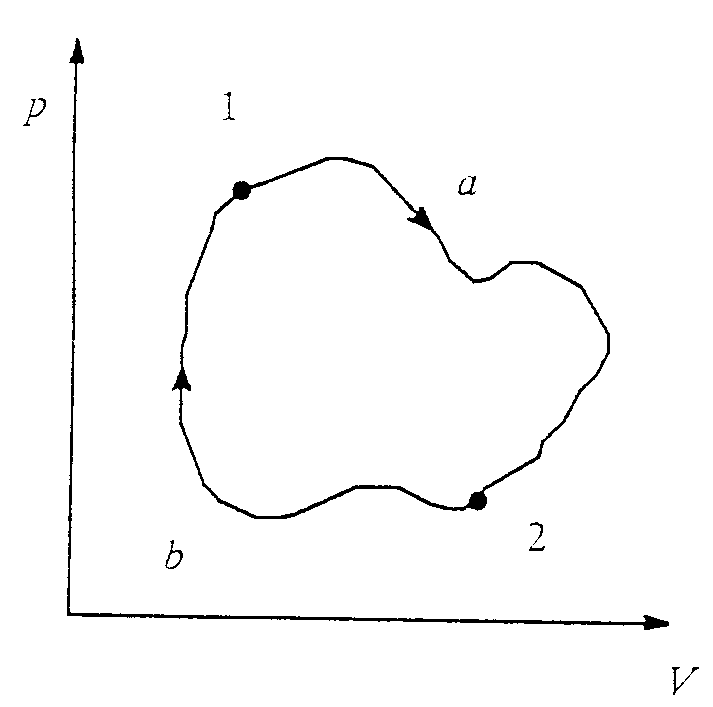
das Differential einer Größe ist, die nur vom Zustand des betreffenden Systems abhängt, also eine Zustandsgröße ist. Das obige Integral kann man auch als Summe zweier Teilintegrale darstellen, die die Integralwerte bei Integration vom Zustand 1 über den Prozeßweg (a) bis zum Zustand 2, und von 2 über den Weg (b) zurück nach 1 wiedergeben.
Dann gilt mit
und
Damit ist bewiesen, daß es sich bei der Entropie um eine Zustandsgröße handelt, da eine Zustandsänderung unabhängig vom Weg ist. Bei der reversiblen Zustandsänderung vom Zustand 1 zum Zustand 2 gilt somit für die Änderung der Entropie des Systems
Die Entropie S ist neben der inneren Energie U die wichtigste Zustandsfunktion eines Systems. Diese beiden Zustandsgrößen bestimmen zusammen das thermodynamische Verhalten des Systems. Wie bei der inneren Energie ist die Definition der Entropie nur bis auf eine additive Konstante festgelegt, da hier ebenfalls nur Aussagen über die Änderung der Entropie bei einer Zustandsänderung gemacht werden.
Der Nullpunkt ist somit willkürlich definierbar.
Bei der Berechnung der Entropieänderung muß immer ein im Prinzip realisierbarer reversibler Weg beschritten werden. Bei adiabatischen Zustandsänderungen, die reversibel ablaufen, ist d Qrev = 0. Somit gibt es keine Entropieänderung (S1 = S2).
Merke: Bei adiabatischen Zustandsänderungen, die reversibel ablaufen, ist d Qrev = 0.
Es gibt keine Entropieänderung: S1 = S2. |
Bei einer Zustandsänderung eines idealen Gases läßt sich die Entropieänderung mit Hilfe des I. Hauptsatzes berechnen:
Aus
folgt mit dem I.Hauptsatz
Mit
folgt dann
Mit
folgt
und mit
gilt schließlich
Setzt man nun voraus, daß die molare Wärmekapazität CV = const kann integriert werden kann, so errechnet man durch Integration ![]()
Wird die innere Energie U durch die Enthalpie H ausgedrückt, erhält man
damit folgt
Vorsicht: Diese Formeln kann man nur anwenden, wenn der Prozeß reversibel ist, aber nicht bei irreversiblen Prozessen. Dort ist
2. Schritt: Wie berechnet man die Entropieänderung dS bei einem irreversiblen Prozeß ?
Wie sieht also der Zusammenhang zwischen D S und irreversiblen Prozessen aus? Zur Beantwortung dieser Frage betrachten wir einen Carnotschen Kreisprozeß, der irreversible Anteile hat. Diese können z.B. Wärmeverluste sein.
Dann ist der Wirkungsgrad geringer als bei vollkommener reversibler Prozeßführung:
mit
und
folgt
und
Dann gilt wegen h irrev < h rev
Mit dem Wirkungsgrad m rev
und dem Wirkungsgrad m irrev
folgt daraus
Somit gilt für beliebige irreversible Kreisprozesse mit der gleichen Beweisführung wie für reversible Prozesse:
Allgemein gilt dann für beliebige Kreisprozesse
Das Gleichheitszeichen bezieht sich hierbei nur auf reversible Kreisprozesse.
Um allgemeine Aussagen über Entropieänderungen zu bekommen betrachten wir nun einen Kreisprozeß, der aus einem irreversiblen (1 ® 2) und einem reversiblen (2 ® 1) Weg besteht. Der gesamte Prozeß ist damit irreversibel. Dann gilt
Mit
und
![]()
folgt
Damit gilt
Für den allgemeinen Fall (Qrev oder Qirrev) folgt:
Das Gleichheitszeichen trifft dabei nur bei reversiblen Prozessen zu.
Für ein abgeschlossenes System ergibt sich
mit
1) Mischen von Wasser
In diesem Beispiel berechnen wir die Entropiedifferenz der folgenden Situation: Je ein Kilogramm 100░C und 0░C warmes Wasser werden gemischt. Die Temperatur des gemischten Wasser beträgt dann 50░C. Es ist jedoch ohne Eingriff von außen nicht möglich, zwei Kilogramm 50░C warmes Wasser in je ein Kilogramm 100░C und 0░C warmes Wasser zu trennen.
Nach der Definition der Entropie gilt
mit d Q = m cp dT folgt
Nun definieren wir den Nullpunkt
Mit
folgt
und
Dann folgt aus
2) Freie, adiabatische Expansion eines idealen Gases
In diesem Beispiel berechnen wir die Entropiedifferenz bei einer freien adiabatischen Expansion eines idealen Gases von V1 auf V2. Dabei werde das Volumen verdoppelt, d.h. es gelte
Mit dem Attribut "frei" bezeichnen wir wieder eine Expansion, bei der weder Wärme zugeführt, noch Arbeit am System geleistet wird
Als Gedankenexperiment zu dieser Rechnung betrachte man zwei gleich große Volumina V1 und V2, von denen eins mit einem idealen Gas, das andere hingegen mit Vakuum gefüllt sei. Durch ein Ventil, das im Grundzustand geschlossen sei, können die beiden Volumina verbunden werden. Dann expandiert das Gas frei in ein Volumen V2 = 2 V1.
Wie groß ist nun D S ?
Zur Berechnung der Entropieänderung können wir nicht die Formel dS = d Q / T verwenden, da es sich um einen irreversiblen Prozeß handelt, denn nur durch zusätzliche Arbeitsleistung kann dieser Prozeß rückgängig gemacht werden.
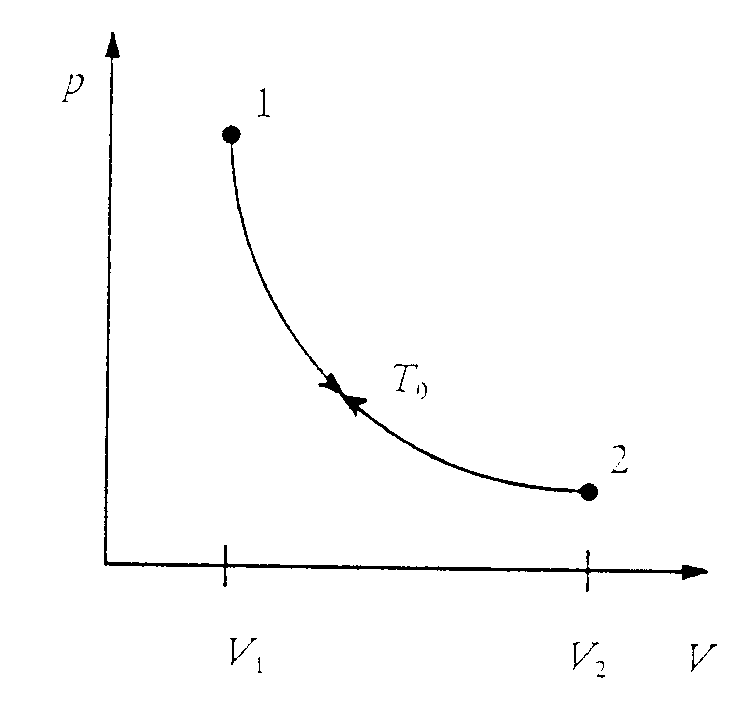
Als Ausweg suchen wir wie oben einen reversiblen Weg, der die Expansion wieder rückgängig macht. Hierzu wählen wir einen isothermen Prozeß. Dann erreichen wir den Anfangszustand wieder, denn der erste Prozeß war ebenfalls bei idealem Gas isotherm.
Die Zustandsänderung besteht dann aus zwei Schritten:
1. Adiabatische (isotherme) Expansion von V1 auf 2V1 bei T0 = const.
2. Isotherme Kompression von 2V1 auf V1 bei T0 = const.
Die Entropie über den Gesamtweg beträgt dann null:
Mit der bereits üblichen Notation folgt daraus
Da die Zustandsänderung isotherm bei T0 verläuft, gilt ebenfalls
Mit der für isotrherme Zustandsänderungen hergeleiteten Formel
gilt dann
Mit V2 = 2 V1
folgt
Für D S12 folgt mit D S12 = - D S21
Wir halten fest, daß bei freier Expansion und anschließender isothermer Kompression gilt:
aber für
gilt mit d Q = 0
und mit
gilt
Damit ist
im Einklang mit dem II. Hauptsatz der Wärmelehre.
Also gilt
Bei einem Kreisprozeß kann die geleistete Arbeit pro Zyklus abgelesen werden als Differenz der 'Arbeitsflächen' der einzelnen Schritte.
Analog kann man ein T/S-Diagramm betrachten. Aus der Definitionsgleichung der Entropiedifferenz
folgt, daß in einem T/S-Diagramm die reversibel umgesetzte Wärmemenge als Fläche unter der Kurve einer Zustandsänderung abgelesen werden kann. Auch hier wird die nach Durchlaufen eines Kreisprozesses erzeugte Wärme angegeben durch die Diffenerenz der 'Wärmeflächen' in den einzelnen Schritte.
Diese Abbildung zeigt das Wärmeschaubild des Carnot-Prozesses. Hierbei entspricht die zugeführte Wärme der Fläche unterhalb der Geraden 3 - 4. Die abgegebene Wärme ist sichtbar unter der Geraden 1 - 2. Die Nutzarbeit entspricht wie beim p,V-Diagramm dem Flächeninhalt der umfahrenen Figur.
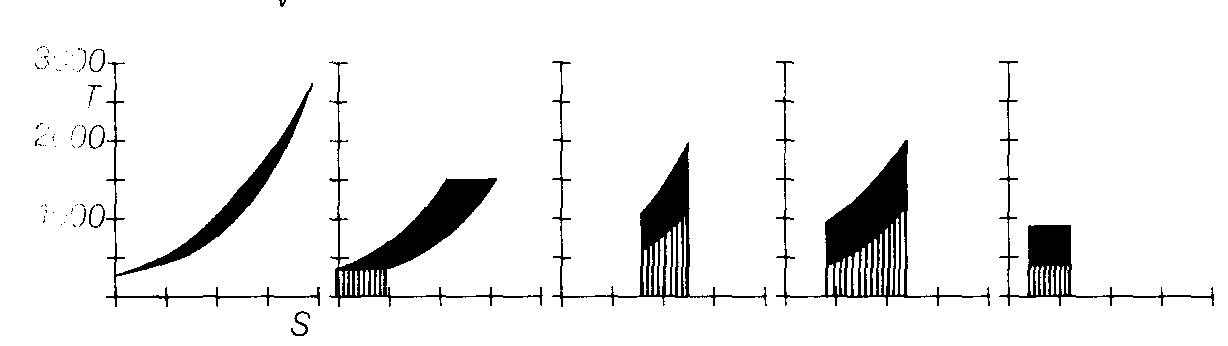 Die Entropieänderung ergibt somit die Größe der mechanischen Arbeit, die sich bei isothermer Ausdehnung bestenfalls gewinnen läßt. Für das ideale Gas ergibt der Energiesatz Q = W.
Die Entropieänderung ergibt somit die Größe der mechanischen Arbeit, die sich bei isothermer Ausdehnung bestenfalls gewinnen läßt. Für das ideale Gas ergibt der Energiesatz Q = W.
Mit dem II. Hauptsatz
folgt
Das Gleichheitszeichen für nur für reversible Ausdehnung.
Merke: Die maximale Arbeit, die sich gewinnen läßt ist gegeben durch W Ż T D S. |
Merke: Der I. Hauptsatz bestimmt, welche Prozesse in einem isolierten System möglich sind.
Der II. Hauptsatz gibt an, in welcher Richtung diese Prozesse ablaufen. |
Merke: Bei jeder Zustandsänderung eines isolierten Systems nimmt die Entropie S zu oder bleibt bestenfalls konstant. |
Mit dieser Aussage läßt sich nun in Analogie zur Mechanik ein thermodynamisches Gleichgewicht formulieren:
Merke: Isolierte Systeme befinden sich im stabilen thermodynamischen Gleichgewicht, wenn ihre Entropie ein Maximum aufweist. Wird also das System gestört, so entwickelt es innere Mechanismen, die es wieder in Richtung maximaler Entropie treiben. |
Es gelten folgende Gleichgewichtsbedingungen:
D
S bezeichnet dabei die Änderung im betrachteten System und D Q bezeichnet den Wärmeaustausch mit der Umgebung.Äquivalent sind die Aussagen
und
Merke: Die Gleichgewichtsbedingung für ein thermodynamisches Gleichgewicht ist 0 = D U - D W - T D S . |
Was genau die Entropie S aussagt und welche Bedeutung dem Entropiebegriff in der Thermodynamik zukommt, wurde vor allem von
![]() L. Boltzmann
aufgeklärt. Die Erkenntnisse, die hierbei von ihm gewonnen wurden, zeigen erst die eigentliche Bedeutung der Entropiebegriffes, der im Rahmen der phänomenologischen Wärmelehre zunächst als recht abstrakte Größe erscheint.
L. Boltzmann
aufgeklärt. Die Erkenntnisse, die hierbei von ihm gewonnen wurden, zeigen erst die eigentliche Bedeutung der Entropiebegriffes, der im Rahmen der phänomenologischen Wärmelehre zunächst als recht abstrakte Größe erscheint.
Um den Begriff der Entropie zu erläutern, betrachten wir wieder die freie Expansion eines Gases von V2 auf V1 in einem abgeschlossenen System. Dieser Prozeß ist selbstablaufend und nach dem II. Hauptsatz nimmt die Entropie des Gases wie in XIV.8.3 berechnet zu, bis die Entropie den unter den neuen Bedingungen möglichen Maximalwert erlangt hat. Das Gas liegt dann nach Kapitel XIV.8.4 wieder in einem Gleichgewichtszustand vor.
Ein Gleichgewichtszustand bedeutet eine gleichmäßige Verteilung der Moleküle auf das Volumen V1. Gehen wir hierbei von einem idealen Gas aus, so hat sich die Entropie bei diesem Prozeß insgesamt um
vergrößert. Das Gas kann von selbst nicht mehr aus diesem Gleichgewichtszustand in andere Zustände übergehen, da dies eine Entropieverminderung zur Folge hätte. Das ist nach dem II. Hauptsatz für ein abgeschlossenes System unmöglich.
Ziel dieses Kapitels ist es nun, den Zusammenhang zwischen Entropie und der Wahrscheinlichkeit quantitativ zu erfassen. Hierbei ist jedoch nicht die statistische Wahrscheinlichkeit w1 gemeint, sondern die in der Thermodynamik zur Kennzeichnung der Zustandswahrscheinlichkeit benutzte Größe Pi, die thermodynamische Wahrscheinlichkeit genannt wird.
Hierzu wollen wir nun eine Erklärung mit Hilfe von statistischen Betrachtungen geben und nehmen dazu ein Gas mit
Moleküle in V1, das sich im thermodynamischen Gleichgewicht befindet, d.h. die Moleküle sind im Zustand 1 gleichmäßig auf das Volumen V1 verteilt.
Wir zeigen nun, daß es extrem unwahrscheinlich ist, daß sich alle Moleküle zufällig wieder in V2 < V1 befinden (Zustand 2). Die Wahrscheinlichkeit![]() , sich im Volumen V2 aufzuhalten, ist für jedes Teilchen gleich groß, und zwar
, sich im Volumen V2 aufzuhalten, ist für jedes Teilchen gleich groß, und zwar
Somit ist die Wahrscheinlichkeit, ein bestimmtes Teilchen in V2 anzutreffen
Die Wahrscheinlichkeit dafür, daß zwei unabhängige Teilchen sich gleichzeitig in V2 befinden, ist dann gleich dem Produkt der Einzelwahrscheinlichkeiten, also
Bei drei Teilchen ist
usw., d.h. die Wahrscheinlichkeit dafür, alle N Teilchen in V2 anzutreffen ist
Ist nun P1 die thermodynamische Wahrscheinlichkeit für den Zustand 1, dann gilt für die thermodynamische Wahrscheinlichkeit des Zustandes 2
Das thermodynamische Wahrscheinlichkeitsverhältnis W ist dann
Dieses Verhältnis gibt an, um wieviel der Zustand 1 wahrscheinlicher ist als der Zustand 2.
Merke: Das thermodynamische Wahrscheinlichkeitsverhältnis ist
W = |
Da
und N immer eine sehr große Zahl ist, z.B. bei n = 1 Mol
ist
Es ist also äußerst unwahrscheinlich, daß sich das Gas irgendwann von selbst auf ein kleineres Volumen zurückzieht.
Um zu Zahlen vernünftiger Größenordnung zu gelangen, bestimmt man den natürlichen Logarithmus des Wahrscheinlichkeitsverhältnisses:
Soll nun die Entropie ein Maß für die Zustandswahrscheinlichkeit darstellen, so muß ein funktionaler Zusammenhang
existieren, d.h. ein Zusammenhang mit der Entropiedifferenz zwischen den Zuständen 1 und 2. Die Änderung der Entropie vom Zustand 1 zum Zustand 2 kann mit Hilfe eines reversiblen Ersatzprozesses berechnet werden, der z.B. isotherm abläuft. Die Entropieänderung ist dann
Für n = 1 Mol gilt mit
Mit ln W = N ln ![]() folgt
folgt
Dieser Vergleich zeigt den von L. Boltzmann gefundenen Zusammenhang zwischen Entropieänderung und thermodynamischem Wahrscheinlichkeitsverhältnis
lFür die Entropie selbst gilt
Merke: Der Zusammenhang zwischen Entropie und Wahrscheinlichkeit ist gegeben durch ln W = |
Merke: Die Entropie eines Systems um so höher ist, je größer die Wahrscheinlichkeit ist, mit welcher der Zustand des Systems realisiert werden kann.
|
Merke: Die Entropie eines Systemzustandes ist proportional dem Logarithmus seiner Wahrscheinlichkeit.
|
Um abschließend noch einen weiteren Aspekt der Entropie darzustellen, betrachten wir folgendes Beispiel:
In einem Behälter befinden sich zwei Gase (Zustand 1), die durch eine Trennwand von-einander getrennt sind. Entfernt man die Trennwand, diffundieren die Gase (Zustand 2). Dieser typische irreversible Mischvorgang kann vom Standpunkt der Wahrscheinlichkeitsrechnung so interpretiert werden, daß das System vom unwahrscheinlichen Zustand hoher Ordnung (Zustand 1) in den wahrscheinlicheren Zustand großer Unordnung (Zustand 2) übergeht, d.h., daß von selbst ablaufende Vorgänge stets von geordneten Zuständen in Richtung größerer Unordnung gehen. Da sie stets mit einem Entropieanstieg verbunden sind, folgt:
Merke: Die Entropie ist ein Maß für den Grad der Unordnung eines Systems.
|